Updated : Thu, October 14, 2021 @ 10:01 PM
SARS-CoV-2 mutations and associated new variants are being reported regularly in the media and scientific literature. The rapid increase in the rate of infection in India was partly attributed to the emergence of three related variants: Kappa (B1.617.1), Delta (B1.617.2) and B1.617.3. In September 2021, the Delta variant is dominant in much of the world.1 Most of the reported mutations affect the Spike-gene, but there are also reported mutations along the SARS-CoV-2 gRNA. As previously reported (Technical Bulletin 1), LGC, Biosearch Technologies has a four-tier assay screening and replacement programme to identify and mitigate against the risk of assay impairment in the event of a mutation arising within the target binding site of diagnostic assays.
- Assays are monitored at fortnightly intervals. This includes the N1 and N2 assays which are components of our ultra-high-throughput SARS-CoV-2 testing systems, 2019-nCoV ValuPanel Reagents™, and the SC2 assay (N gene) which is a component of the Influenza SARS-CoV-2 Multiplex ValuPanel Reagents (Table 1).
- Each of these sequences is compared to published sequences that are reported in the GISAID database.
- Any mutations of concern are subjected to further in silico analysis and wet lab verification if required.
- Additional assays have been evaluated as ready for deployment, should a detrimental mutation be identified. These additional assays are also under constant surveillance.
Table 1 Sequences of oligonucleotides used in SARS-CoV-2 detection assays (written in 5′ to 3′ direction)
| Assay |
Oligo ID |
Sequence |
| CDC N1 |
2019-nCoV_N1-F |
GACCCCAAAATCAGCGAAAT |
| 2019-nCoV_N1-R (RT primer) |
TCTGGTTACTGCCAGTTGAATCTG |
|
| 2019-nCoV_N1 Probe |
FAM-ACCCCGCATTACGTTTGGTGGACC-BHQ-1 |
|
| CDC N2 |
2019-nCoV_N2-F |
TTACAAACATTGGCCGCAAA |
| 2019-nCoV_N2-R (RT primer) |
GCGCGACATTCCGAAGAA |
|
| 2019-nCoV_N2 Probe |
FAM-ACAATTTGCCCCCAGCGCTTCAG-BHQ-1 |
|
| RNase P |
RP-F |
AGATTTGGACCTGCGAGCG |
| RP-R |
GAGCGGCTGTCTCCACAAGT |
|
| RP-P (CFO560) |
CalFluorOrange560-TTCTGACCTGAAGGCTCTGCGCG-BHQ-1 |
|
| SC2 |
SC2-F |
CTGCAGATTTGGATGATTTCTCC |
| SC2-R |
CCTTGTGTGGTCTGCATGAGTTTAGG | |
| SC2-P (CFR610) | CalFluorRed610ATTGCAACAATCCATGAGCAGTGCTGACTC-BHQ1 |
Mutation analysis procedure
- Fortnightly in silico screen evaluation of sequences submitted to GISAID: N1, N2, SC2 and variant specific ValuPanel (see Technical Bulletin 2) assay oligonucleotide (oligos) target sequences are screened against SARS-CoV-2 gRNA sequences submitted to the GISAID SARS-CoV-2 database. Mismatches between database viral sequence and assay oligonucleotide sequences are highlighted for further investigation.
- News reports, customer enquiries, public health, government technical briefing documents: Any mutations reported in news outlets, reported to Biosearch Technologies by customers or published in official communications such as technical briefings (PHE), WHO or CDC communications are also highlighted for further investigation.
- Screening of N1/N2 replacement assays: There is a programme in place to identify assays that could mitigate for sub-optimal performance when using the N1 or N2 (Table 1) assays, should a mutation occur in the target sites which cause assay failure (see step 4). These potential mitigation assays have undergone thorough sequence evaluation and are also included in the regular screen against sequences held in GISAID. If any mutation lies in the target site of the potential mitigation assays, these are also highlighted for further investigation.
Mutation analysis and investigation
Prevalence:
- All mutations occurring in at least 0.1% of GISAID global sequences and lying in the target sequences of the N1, N2, SC2 and mitigation assays are investigated.
- All mutations lying in the target sequences of the N1, N2, SC2 and mitigation assays occurring in at least 1% of sequences from an individual country are also examined.
Mutation location: Of the sequences with the defined prevalence in GISAID, mutations lying within the five bases at the 3′ of a primer or the central 50% of a probe are taken forward for wet lab testing. Single base mismatches lying outside of this region are reported to have low impact on assay performance.2
Tm change estimate: The Tm of the assay oligo with the matched target sequence is compared to that of the mismatched target using in silico assessment.
In silico screening results
The N1, N2, SC2 and mitigation assay sequences were screened against sequences submitted to GISAID (up until September 2021). No mutations of concern were identified in the potential mitigation assays or in SC2. Eight mutations in N1 and N2 assay oligo target sites were highlighted for further investigation and for wet lab analysis.
With the exception of T29194C, the predicted Tm differences were suggestive of a potentially detrimental effect of the mutation on the assay (Table 2). Each of the mutations was then taken into a wet lab testing protocol.
Table 2 Analysis of mutations of concern identified in N1 or N2 assays
Mutations are relative to the SARS-CoV-2 reference sequence NC_045512.2 and the naming convention is described in “Emerging SARS-CoV-2 mutations and their effect on the immune response” technical bulletin.3
| Mutation in probe central region or primer 3' (five bases) |
Assay affected |
Oligo |
Frequency of mutation (GISAID global) |
Delta Tm (°C) |
Percentage mismatch |
| G28321A |
N1 |
probe |
0.094 |
-3.7 |
4.2 |
| G28321T |
N1 |
probe |
0.11 |
-3.7 |
4.2 |
| G28300C |
N1 |
F primer |
0.04 |
-3.5 |
5 |
| G28300T |
N1 |
F primer |
0.28 |
-4.3 |
5 |
| G29179T |
N2 |
F primer |
0.54 |
-6.6 |
5 |
| C29197T |
N2 |
probe |
1 |
-5.8 |
5 |
| T29194C |
N2 |
probe |
0.25 |
-1.9 |
4.35 |
| C29200T |
N2 |
probe |
0.21 |
-5.6 |
4.35 |
Wet lab testing results
In the absence of viral templates for these mutations, a template DNA amplicon containing the mutation was synthesised, along with wild-type templates. The concentration of each synthetic template was normalised to that of the AccuPlex™ SARS-CoV-2 Reference Material. All targets were detected using the ultra-high-throughput SARS-CoV-2 N1/N2/RnP assay, RapiDxFire™ qPCR 5X Master Mix GF, including EpiScript™ Reverse Transcriptase.
The ultra-high-throughput SARS-CoV-2 N1/N2/RnP assay contains assay oligonucleotides to detect SARS-CoV-2 N1 and N2 targets (Table 1) and the human RNase P. The N1 and N2 specific probes are both labelled with FAM and the Human RNase P detection is from a CalFluor™ Orange 560 (CFO560) labelled probe.
End-point analysis:
End-point analysis was performed using the Nexar™, Hydrocycler2™ and Araya™ platforms.
This high-throughput workflow supports the analysis of 150,000 samples per day. Reactions were prepared in the Nexar liquid handling system and dispensed into wells of the Array Tape™ consumable. The total reaction volume was 5 µL, of which 3.8 µL was the sample and the remainder RT-PCR Master Mix. Samples were included at 500, 50 and 5 copies per reaction and 10 replicates run for each concentration. Fifty PCR cycles were run in the Hydrocycler2 and then the fluorescence was read using the Araya plate reader.
For each of the eight mutations analysed, the mutated and wild-type templates could not be differentiated when 500, 50 or 5 copies were included in the reaction and end-point fluorescence was detected. Based on this data, these mutations have not been highlighted as a risk to the continued use of the ultra-high-throughput SARS-CoV-2 N1/N2/RnP assay (ePCR) (Table 3).
Real-time analysis
Real-time analysis was carried out using a Bio-Rad CFX. This is a relatively low-throughput system using larger volume samples in a 96-well format. For this study, 20 µL reaction volumes contained 500 copies of the template carrying the specific mutation of interest or the wild-type construct.
The impact of primer and probe mutations will depend on the nature of the nucleotide mismatch and position within the oligonucleotide. Previous studies of melting curve analysis using HyBeacon probes indicated that destabilising mismatches (C/A, C/T and C/C) will yield ΔTms of 8-10 °C, intermediate mismatches (A/A, A/C, T/T, and T/C) will yield ΔTms of 6-8 °C and stable mismatches (A/G, T/G, G/A, G/T and G/G) will result in ΔTms of 4-6 °C. The impact of a mutation depends also on context and location. In addition, the impact on the overall assay depends also on which oligonucleotide is affected. Mutations in the primer serving as the reverse transcription primer impact the initial generation of cDNA, thus reducing template in the reaction. Mutations in either primer target sequence may result in inefficient priming and initially delay PCR. However, each amplicon then contains the primer sequence facilitating subsequent efficient PCR. Mutations within the probe result in inefficient binding and cleavage of the probe at each cycle, thus, increasing Cq indirectly when the ΔRFU is reduced. It is predicted that mutations located towards oligonucleotide ends will reduce probe stability to a lesser extent. However, mismatches located towards the 3´ ends of primers will have a greater impact on assay efficiency.
The impact of mutations was measured by comparison of amplification of each mutation-containing construct to the amplification of the wild-type target (N1 WT SPUD, N2_WT_SPUD) and the AccuPlex SARS-CoV-2 package virus control material using measures of both Cq (Figure 1) and end-point relative fluorescence (Figure 2). Statistical analysis of these data was used to identify mutations that resulted in deviations from the wild-type.
Under these conditions, the four N1 mutations resulted in up to one cycle delay when compared to the wild-type construct. Two of the N2 mutations G29179T, T29194C also resulted in less than one cycle delay. There was a slightly higher delay of around two cycles in the presence of the C29197T and C29200T (Figure 1).
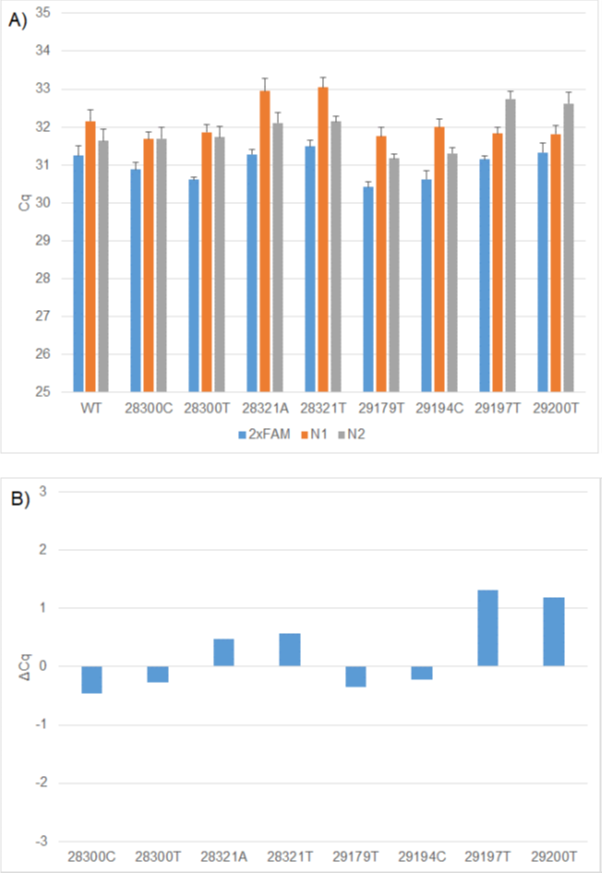
Figure 1: A) Comparison of WT and mutation Cq values using 2x FAM (N1 and N2), 1x FAM (N1) and 1x FAM (N2) RT-PCR formulations. B) ΔCq values between WT and mutation constructs using the relevant 1x FAM formulation. The N1 WT, N2 WT, RP WT replicates generated on individual CFX plates were used to calculate the ΔCq's for each mutation construct.
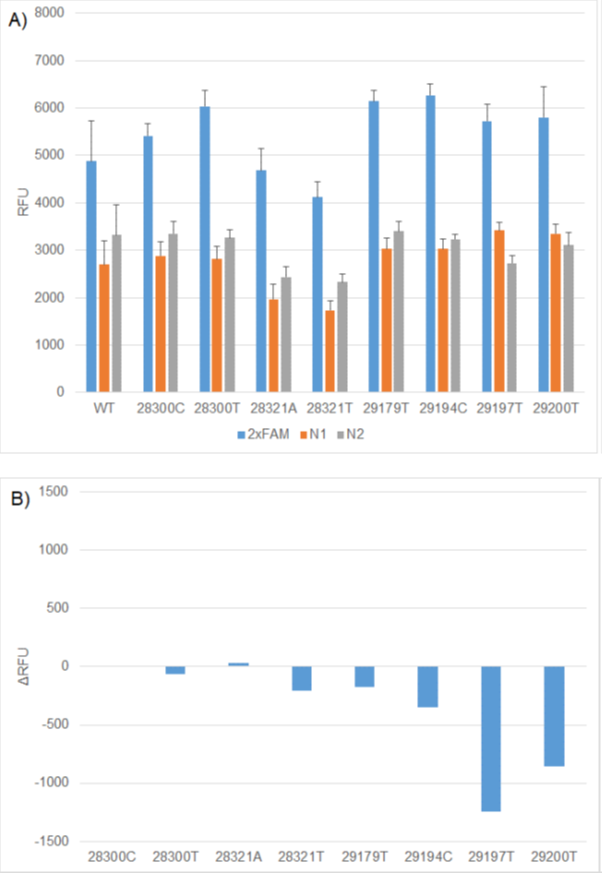
Figure 2. A) Comparison of WT and mutation RFU values using 2x FAM (N1 & N2), 1x FAM (N1) and 1x FAM (N2) RT-PCR formulations. B) ΔRFU values between WT and mutation constructs using the relevant 1x FAM formulation. The N1 WT, N2 WT, RP WT replicates generated on individual CFX plates was used to calculate the ΔRFUs for each mutation construct.
Table 3 Analysis of mismatches due to mutations
| Mutation in probe central region or primer 3' (five bases) |
Assay affected |
Oligo |
Frequency of mutation (GISAID global) |
Delta Tm (°C) |
Percentage mismatch |
Change in sensitivity of target detection RT-qPCR (ΔCq relative to wild-type) |
Change in sensitivity of target detection ePCR |
| G28321A |
N1 |
probe |
0.094 |
-3.7 |
4.2 |
1 |
0 |
| G28321T |
N1 |
probe |
0.11 |
-3.7 |
4.2 |
<1 |
0 |
| G28300C |
N1 |
F primer |
0.04 |
-3.5 |
5 |
<1 |
0 |
| G28300T |
N1 |
F primer |
0.28 |
-4.3 |
5 |
<1 |
0 |
| G29179T |
N2 |
F primer |
0.54 |
-6.6 |
5 |
<1 |
0 |
| C29197T |
N2 |
probe |
1 |
-5.8 |
5 |
2 |
0 |
| T29194C |
N2 |
probe |
0.25 |
-1.9 |
4.35 |
<1 |
0 |
| C29200T |
N2 |
probe |
0.21 |
-5.6 |
4.35 |
<2 |
0 |
There was no absolute relationship between the predicted change in Tm or percentage mismatch and the result of functional testing. As predicted, the mismatches with C in the probe had more impact than other probe mutations or primer mutations. However, the two N2 mutations within the probe resulted in the second and third highest Tm differences. One of these is C29200T. Ziegler et al, (2020) reported that this mutation had little impact on assay performance under their conditions, as observed in this study.4 This data illustrates that the wet lab analysis is critical for understanding the impact of emerging mutations on diagnostic assay performance.
Conclusion
Biosearch Technologies has adopted an aggressive screening approach of our diagnostic assays to ensure the highest vigilance. Regular in silico analysis of mutations of concern ensures the rapid identification of any potential lack of function of our assays. Subsequent wet lab testing facilitates an understanding of how any mutation would impact the assay in the diagnostic setting. The mitigation assay programme is in place to swiftly deploy a new assay should we discover that the sequences of the assay are compromised.
References
- Overview of Variants in Countries. https://covariants.org/per-country. Accessed September 2021.
- Lefever S, Pattyn F, Hellemans J, Vandesompele J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin Chem. 2013 Oct;59(10):1470-80. https://doi.org/10.1373/clinchem.2013.203653. Epub 06 Sep 2013.
- Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome. NCBI Reference Sequence: NC_045512.2. https://www.ncbi.nlm.nih.gov/nuccore/NC_045512.
- Ziegler K, Steininger P, Ziegler R, Steinmann J, Korn K, Ensser A. SARS-CoV-2 samples may escape detection because of a single point mutation in the N gene. Euro Surveill. 2020;25(39):pii=2001650. https://doi.org/10.2807/1560-7917.ES.2020.25.39.2001650. 30 Sep 2020.
