Updated : Thu, December 16, 2021 @ 6:46 PM
Emerging SARS-CoV-2 variants of concern and variants of interest are defined by agencies such as the World Health Organisation (WHO). Such variants are recognised by lineage defining mutations, which are prompting scientists to monitor any that may be advantageous in escaping an immune response. This requires vigilant screening of viral genomic sequences, enabled by genomic tools to ensure SARS-CoV-2 vaccines remain efficacious. In this technical bulletin, we review the immunologic impact of SARS-CoV-2 lineage defining mutations in the Delta and Lambda variants.
The initial publication of the SARS-CoV-2 sequence1,2 was based on sequencing of viral material isolated from clinical samples extracted from patients in Wuhan, China who were experiencing a novel respiratory syndrome, now referred to as COVID-19. Since the original SARS-CoV-2 sequence was published, several different variants and strains have been described.3 WHO has named a number of these according to the Greek alphabet (Table 1).
Table 1 WHO assigned variant nomenclature
Conventionally, variants are referred to as viruses with genomes that differ from each other due to mutations (Figure 1). The Alpha variant lineage (B1.1.7; Table 1) was first defined in samples in Kent, UK, in September 2020. The Alpha variant differs from the original Wuhan genome by 17 lineage defining mutations4, which include deletion of amino acid Y144 (genomic location 21991-21993 in reference sequence NC_045512) and a single nucleotide polymorphism (SNP; C23604A) in the spike protein resulting in the P681H mutation (Table 2). The Alpha variant spread rapidly across Europe to become the dominant strain and has since been identified across the world.5
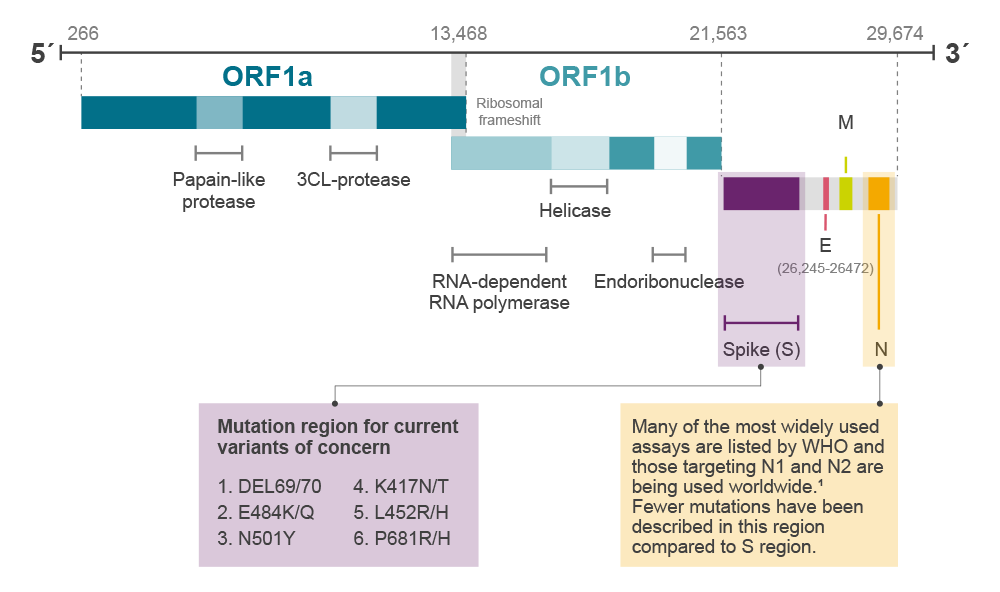
Figure 1. gRNA and gene arrangement of SARS-CoV-2 genome
More recently, spread of the Delta variant (B.1.617.2, originally described in a traveller from India) has been seen to be rapidly overtaking Alpha.6 The Delta variant is one of a family of three similar viruses (along with Kappa, B.1.617.1 and B.1.617.3). All three of the B.1.6177 variants have acquired L452R and P681R mutations in the Spike gene, along with V82A in ORF7a and R203M in the N gene. L452R was first described in the Epsilon variants, circulating in California, USA. The Delta variant is differentiated from B.1.617.1 and Kappa in that it also carries the T478K Spike mutation and is wild type (E) at Spike 484. Both the B.1.617.1 and Kappa have acquired the E484Q mutation (Table 2). E484Q results from a G to C mutation at genome location 23012, whereas the previously described E484K is caused by a G to A mutation at the same site. E484K is a defining mutation for Beta, Gamma, Eta, Theta and Iota lineages. The Delta variant continues to acquire mutations, with sequences being described containing Y144 deletion as seen in Alpha variant, and the K417N mutation (this is referred to as Delta plus)8, previously associated with the Beta variant.
The Lambda variant, prevalent in South America and recently discovered in Brazil, is now designated as a variant of interest by the World Health Organisation (WHO; 14 June 2021).9 This carries different mutations but at the same site as some of those reported for Delta and Alpha. For example, the Lambda spike location L452 has mutated to 452Q, and the N-gene R203 mutation is to lysine (R203K) rather than the methionine (R203M) seen in the Delta variant.
Table 2 Differentiating mutations for identification of SARS-CoV-2 Alpha and Delta strains
|
Alpha |
Kappa |
Delta |
|
Lambda |
||
|
Spike protein mutation |
Genome mutation |
B1.1.7 |
B1.617.1 |
B1.617.2 |
B1.617.3 |
C.37 |
|
L452R L452Q |
T22917G T22917A |
L |
R |
R |
R |
Q |
|
P681H P681R |
C23604A C23604G |
H |
R |
R |
R |
P |
|
E484K E484Q |
G23012A G23012C |
E |
Q |
E |
Q |
E |
|
T478K |
C22995A |
T |
T |
K |
T |
T |
|
T19R |
C21618G |
T |
T |
R |
R |
T |
|
F490S |
T23031C |
F |
F |
F |
F |
S |
|
G75V |
G21786T |
G |
G |
G |
G |
V |
|
Δ246-252 |
22298-22318D |
- |
- |
- |
- |
Δ |
|
T859N |
C24138A |
T |
T |
T |
T |
N |
|
T76I |
C21789T |
T |
T |
T |
T |
I |
|
ORF1a mutation |
|
|
|
|
|
|
|
Δ3675-3677 |
11288-11296D |
- |
- |
- |
- |
Δ |
|
ORF7a mutation |
|
|
|
|
|
|
|
V82A |
T27638C |
V |
A |
A |
A |
V |
|
N gene |
|
|
|
|
|
|
|
R203K R203M |
G28881A G28881T |
R |
M |
M |
M |
K |
When genomic differences confer a difference in phenotype they are referred to as different strains. The phenotypic differences that differentiate strains include differences in viral replication, transmission rates or antigenicity. For the spread of a strain to occur so effectively, despite infection prevention activities in many of the affected countries, is indicative that some of the acquired mutations confer an evolutionary advantage.10,11
One mechanism by which a mutation may confer an advantage is by avoidance of the immune response. The initial immune response to a viral infection is via the B-cell humoral response which is antibody-mediated. Avoidance of humoral immunity is observed as resistance to antibody-mediated neutralisation of SARS-CoV-2, both from convalescent and vaccinated sera. While the Alpha variant has been shown to be sensitive to neutralising antibodies, Beta and Gamma variants are relatively resistant.
The second immune protection against pathogens is cellular immunity. Cellular immunity is cytotoxic T-lymphocyte (CTL) mediated and occurs when CTLs recognise viral epitopes presented on infected cells via human leucocyte antigen (HLA) class 1 molecules. The severity of the resulting COVID-19 resulting from SARS-CoV-2 infection has been inversely correlated to the functionality of cellular immunity.12 Therefore, both humoral and cellular immune responses are involved in protection against SARS-CoV-2 infection. Mutations that facilitate escape from either antibody or cellular recognition would be predicted to confer a survival advantage to the variant.
Immune response to SARS-CoV-2
Evasion of immune responses to SARS-CoV-2 is believed to be conferred primarily by changes in the Spike protein which projects from the surface of the virus.
The immediate immune response to infection is not specific and mediated via cytokines and interferons. Approximately 6-8 days after infection a specific adaptive response is evident. An initial T-cell response is followed by Immunoglobulin M (IgM) and Immunoglobulin G (IgG) antibodies, comprising the adaptive response. Neutralising antibodies bind to the virus and it appears that the most effective neutralisation occurs in the presence of anti-Spike IgG and anti-RBD IgG [RBD; Receptor Binding Domain].13
The RBD lies in the S1 domain of the Spike which interacts with the angiotensin-converting enzyme 2 (ACE-2) receptors, on the surface of the host cells. The lungs are affected by COVID-19 because the ACE-2 receptor is abundant on the surface of type II alveolar cells. Similarly, organs of the digestive system are also targets via ACE-2. Upon binding of the RBD to the ACE-2 receptor, the viral envelope fuses to the host cell membrane. which initialises the process of viral entry into the cell.14
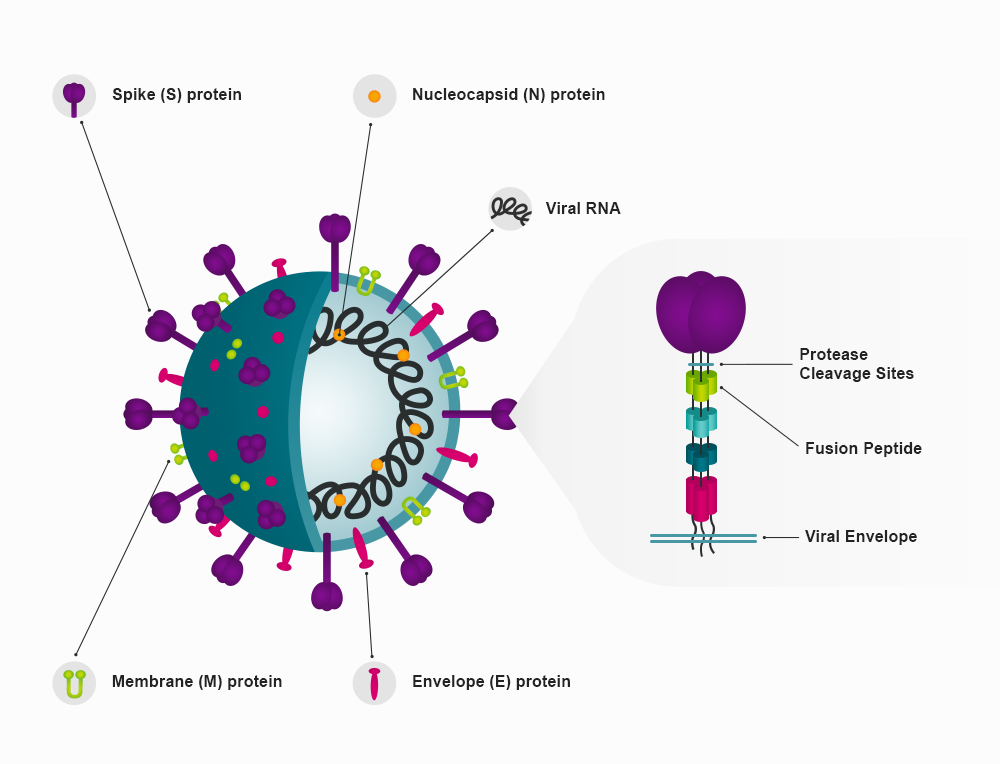
Figure 2. The SARS-CoV-2 spike protein is has a large bulbous region held away from the viral membrane by a slender protein stem. Three of these single spike protein units form a trimer. The bulbous region and the top of the stem from the S1 region, while the S2 region is the remainder is of the stem, which is embedded in the viral envelope.15
The RBD is a looped region formed from spike amino acids 319 to 591. Several mutations have been described within this region, significantly N501Y, T478K, F490S, K417N/T, E484K/Q, and L452R/Q.
The impact of the L452R/Q mutations
The L452R mutation was detected in Denmark (B1.39) in March 2020 and later observed in California,16,17 USA in Epsilon variants (B.1.427 and B.1.429). It has also been described in the strain C.36.3 (VUI-21MAY-02)18 and is a defining mutation in the B.1.617 lineage, which includes the Delta variant. However, it has been identified in sequences not defined as B.1.617 and may be regarded as an adaptive mutation, conferring an evolutionary advantage to those viruses carrying the mutations.19
Structural modelling of the spike protein predicts that L452 forms a hydrophobic contact with L492, which in turn is in a hydrophobic interaction with F490. Together this forms a hydrophobic region on the surface of the RBD in the wild-type strains. The 452R mutation replaces the leucine with the positively charged, hydrophilic arginine, removing the hydrophobic interaction. It is worthy of note that the Lambda variant also carries the F490S mutation, replacing the aromatic phenylalanine with serine.
L452R is a mutation of concern because it enhances viral binding to the ACE-2 receptor, increases viral infectivity and enhances viral replication capacity.20 Vaccine-stimulated antibodies raised to strains carrying the wild-type Leucine are less effective at binding to the 452R altered spike protein.20 There is some evidence that L452R, evades cellular immunity.20 This has been specifically described for HLA-A24 restricted cellular immunity and is relevant to the Delta variant because HLA-A24 is an HLA-1 allele that is predominant in East and South-East Asian populations.20
The Lambda (C.37) variant has recently been designated as a Variant of Interest by WHO.21 This carries the L452Q mutation, which was also feared to impact infectivity or evade previously developed, adaptive immune responses. A pseudotyped lentivirus carrying the L452Q mutation was shown to increase infectivity by two-fold. The L452Q mutation also resulted in a three-fold increase in affinity for the ACE-2 receptor, which is likely to result in increased transmissibility.22 The L452Q mutation conferred a resistance of two to threefold against neutralisation by antibodies in convalescent or vaccine-induced sera, which is similar to that observed for L452R. However, viruses carrying the L452Q were effectively neutralised using the Regeneron monoclonal antibody cocktail. These findings offer hope that current vaccination and therapy protocols would continue to offer effective protection against variants carrying the L452R and L452Q mutations. However, the Lambda and Kappa variants also carry further mutations in the RBD that have also been reported to evade immune responses.
The impact of the E484K/Q mutations
The glutamate (E) to lysine (K) substitution at position 484 (E484K) in the RBD of the spike protein was first described in the Beta and Gamma variants. Reports of the E484K mutation occurred as vaccine programmes were being implemented and raised grave concerns around vaccine effectiveness. Indeed, when the E484K was present in an Alpha variant background, it was more resistant to neutralisation by vaccinated23 and convalescent sera.24,25 In the case of convalescent sera with low IgG against the SARS-CoV-2 spike, there was little, if any, neutralisation of recombinant virus containing E484K.
While the Alpha, Delta and Lambda variants are wild-type at Spike 484 (E484), the Kappa and B1.1.617.3 variants carry the E484Q mutation. E484Q is predicted to result in a disruption of electrostatic bonds involved in the interaction with the ACE-2 receptor. There is also a prediction of a reduction in hydrogen bonds required for binding by the neutralising monoclonal antibody REGN10933 to the RBD, leading to a reduction in neutralisation effectiveness.7
The impact of the P681R/H mutations
After binding to the ACE-2 receptor, fusion of the virion to the host cells requires cleavage of S1 and S2. SARS-CoV-2, like MERS-CoV, has a furin RXXR cleavage site between the S1 and S2 domains which further enhance binding and fusogenicity of the virion to host cells.26 The furin cleavage site is formed from residues 682-685, immediately adjacent to amino acid 681. Two mutations have been described at the Spike 681 amino acid. The Alpha variant carries a genomic C to A mutation such that basic imidazaole-containing histidine replaces the wild-type proline (P681H), optimising the furin cleavage site. In mouse models, the consequences of this mutation include altered fusion kinetics and enhanced infectivity.27
The Delta variant has a C to G mutation resulting in a basic arginine at the same position (P681R). P681R containing variants are highly fusogenic and form prominent syncytia.28 Point mutations of P681R in the original Wuhan lineage did not affect viral infection kinetics or transmissibility suggesting this mutation synergises with other mutations in the spike protein for enhanced pathogenicity.28 Equally important to viral evolution are strategies to countervail COVID-19. P681 is included in the highest-ranked epitope target in silico models for vaccine development.27 Therefore, P681 is predicted to be part of the epitope targeted by the immune response. Lineages harbouring mutations in this region are more likely to challenge the effectiveness of vaccines targeting P681 containing epitopes.
Genotyping to monitor variants in populations
While some incidents of de novo identification of new variants have occurred as the result of the failure of diagnostic tests, for example, in the case of the Alpha variant spike deletion (Δ69/70) or the N-gene D3L, many mutations have been identified through global sequencing projects. However, there is growing interest in using RT-qPCR approaches and genotyping samples to define the variant of interest. This is a rapid and cost-effective approach that facilitates timely monitoring of variant prevalence. Using the rapid genotyping approach allows policymakers to respond to variant changes much faster than when using sequencing. This is one method by which the spread of new variants could be contained. If adopted, and population mixing mitigation action was taken, this approach would also contribute to reducing the development of new variants by reducing the spread of variants into non-vaccinated or partially vaccinated populations.
Genotyping assays for RT-qPCR have been designed to distinguish between the genotypes at locations 484, 452 and 681 (Figure 2). These facilitate rapid identification of the variant under investigation (refer to Table 2). The genotyping assays consist of two primers that are specific to areas of sequence that flank the mutation of interest. In addition, probes are designed that specifically target the wild type or mutation sequence. Each of these probes has a different fluorescent label so that the probe:target match can be identified. During the PCR step, the probe preferentially hybridises to the matched target, resulting in higher end-point fluorescence (or earlier Cq if run in real-time) than that for hybridisation of the mismatch probe to the target. Since many of the mutations under investigation are single nucleotide polymorphisms (SNPs), the probes are shortened by the addition of the BHQplus™ Probes, which also raise the effective annealing temperature. This modification increases the specificity of the matched probe to target sequence. Further modifications have also been incorporated into the assays to ensure functionality when additional mutations occur within the target sequence.
Identification of Delta variant using end-point genotyping
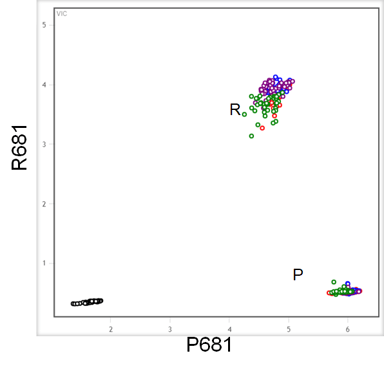
Figure 3. End-point genotyping was used to identify samples containing wild-type or mutated sequences resulting in P or R at amino acid 681. The wild type sequence has a C at position 23604, which is mutated to G in gRNA sequence of the Delta variant. These assays were performed using a high throughput workflow consisting of the Nexar™ liquid handling system, the Hydrocycler2™ to perform PCR cycles and the Araya system for fluorescent label detection.
P681R SARS-CoV-2 Variant ValuPanel™ assay in which the probe for the wild type is labelled with FAM and the mutation with CIV was used to target template included in reactions at 500, 100, 50 and 10 copies of target/reaction. Reactions were run in RT-qPCR performed with RapiDxFire™ qPCR 5X Master Mix with EpiScript™ Reverse Transcriptase (5 U/reaction) and SuperROX (75 nM). Mutation (681R) and Reference strain (P681) were segregated to allele specific clusters in the end-point genotyping experiment such that the Delta variant could clearly be distinguished from any carrying the wild-type genotype.
A call to action for sustained vigilance
The biological, clinical and social impact of mutations within the SARS-CoV-2 genome is becoming more apparent as emerging variants are managed. The rollout of vaccinations may also create evolutionary pressure with further mutations escaping immunity. Management of the pandemic requires a robust genomic surveillance programme that monitors overall case numbers for the continued identification and characterisation of emerging mutations and variants. Learn more about how the team at Biosearch Technologies can support the tracking of variant sequences and enable rapid and cost-effective PCR-based genotyping.
References
- Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome. https://www.ncbi.nlm.nih.gov/nuccore/NC_045512
- Zhang Y-Z. Novel 2019 coronavirus genome. Virological. 2020 Jan 10 [cited 2020 Mar 1]. https://virological.org/t/novel-2019-coronavirus-genome/319
- GISAID Tracking of Variants. https://www.gisaid.org/hcov19-variants/
- Standardised Variant Definitions. https://github.com/phe-genomics/variant_definitions
- B.1.1.7. https://cov-lineages.org/global_report_B.1.1.7.html
- B.1.617.2 https://cov-lineages.org/global_report_B.1.617.2.html
- Cherian, S. et al. Convergent evolution of SARS-CoV-2 spike mutations, L452R, E484Q and P681 R, in the second wave of COVID-19 in Maharashtra, India. bioRxiv 2021.04.22.440932; https://doi.org/10.1101/2021.04.22.440932
- SARS-CoV-2 variants of concern and variants under investigation in England. Technical briefing 16. 18 Jun 2021. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1001359/Variants_of_Concern_VOC_Technical_Briefing_16.pdf
- Wink, P.L. et al. First identification of SARS-CoV-2 Lambda (C.37) variant in Southern Brazil. https://doi.org/10.1101/2021.06.21.21259241
- Duffy, S. et al. Rates of evolutionary change in viruses: patterns and determinants. Nat Rev Genet. 2008 Apr;9(4):267-76. https://doi.org/10.1038/nrg2323. Epub 2008 Mar 4.
- Davies NG, Barnard RC, Jarvis CI, et al. Estimated transmissibility and severity of novel SARS-CoV-2 Variant of Concern 202012/01 in England. CMMID. Preprint published online December 23, 2020. Updated December 31, 2020. https://doi.org/10.1101/2020.12.24.20248822
- Le Bert, N. et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. 2020 Aug;584(7821):457-462. https://doi.org/10.1038/s41586-020-2550-z. Epub 2020 Jul 15.
- Immune responses and immunity to SARS-CoV-2. https://www.ecdc.europa.eu/en/covid-19/latest-evidence/immune-responses
- Sanyal, D. et al. (2020). An Exploration of the SARS-Cov-2 Spike Receptor Binding Domain (RBD) – A Complex Palette of Evolutionary and Structural Features. bioRxiv preprint. https://doi.org/10.1101/2020.05.31.126615.
- Granet, R. The enemy within: How SARS-CoV-2 uses our own proteins to infect our cells. May 21. CAS. https://www.cas.org/resource/blog/covid-19-spike-protein
- Deng, X. et al., Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant. Cell. 2021 Jun 24; 184(13): 3426–3437.e8. Published online 2021 Apr 20. https://doi.org/10.1016/j.cell.2021.04.025
- Zhang, W. et al., Emergence of a Novel SARS-CoV-2 Variant in Southern California. JAMA. 2021;325(13):1324-1326. https://doi.org/10.1001/jama.2021.1612
- phe-genomics / variant_definitions. https://github.com/phe-genomics/variant_definitions/blob/main/variant_yaml/paragraph-footwork.yml
- Sinha Dutta, S. L452R mutation potentially favors adaptive evolution of SARS-CoV-2. 23 Feb 2021. https://www.news-medical.net/news/20210223/L452R-mutation-potentially-favors-adaptive-evolution-of-SARS-CoV-2.aspx
- Motozono, C. et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host & Microbe. Volume 29, Issue 7, 14 July 2021, Pages 1124-1136.e11. https://doi.org/10.1016/j.chom.2021.06.006
- Tracking SARS-CoV-2 variants. World Health Organization. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants
- Tada, T. et al. SARS-CoV-2 Lambda Variant Remains Susceptible to Neutralization by mRNA Vaccine-elicited Antibodies and Convalescent Serum. bioRxiv preprint. https://doi.org/10.1101/2021.07.02.450959.
- Collier D, de Marco A, Ferrerira I, et al. SARS-CoV-2 B.1.1.7 escape from mRNA vaccine-elicited neutralizing antibodies. medRxiv 2021.01.19.21249840v2 [Preprint]. 2021. www.medrxiv.org/content/10.1101/2021.01.19.21249840v2.
- Jangra, S. et al. The E484K mutation in the SARS-CoV-2 spike protein reduces but does not abolish neutralizing activity of human convalescent and post-vaccination sera. medRxiv 2021 Jan 29; 2021.01.26.21250543. https://doi.org/10.1101/2021.01.26.21250543. Preprint
- Jangra, S. et al. SARS-CoV-2 spike E484K mutation reduces antibody neutralisation. The Lancet Microbe. April 07, 2021. https://doi.org/10.1016/S2666-5247(21)00068-9
- Wu, Y., Zhao, S. Furin cleavage sites naturally occur in coronaviruses. Stem Cell Research. Volume 50, Jan 2021, 102115. https://doi.org/10.1016/j.scr.2020.102115
- Maison, D. P. et al. Genetic Characteristics and Phylogeny of 969-bp S Gene Sequence of SARS-CoV-2 from Hawai‘i Reveals the Worldwide Emerging P681H Mutation. Hawaii J Health Soc. Welf. 2021 Mar 1; 80(3): 52-61. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7953241/
- Frazier, L. et al. Spike protein cleavage-activation mediated by the SARS-CoV-2 P681R mutation: a case-study from its first appearance in variant of interest (VOI) A.23.1 identified in Uganda. bioRxiv. 2021 Jul 5;2021.06.30.450632. https://doi.org/10.1101/2021.06.30.450632. Preprint.
Resources
