Updated : Tue, October 11, 2022 @ 8:43 PM
The development of new genomic sequencing techniques and bioinformatics analysis in the last decade has led to massive advances in the diagnosis, prognosis and treatment of disease.1,2
At a time when the sequencing and analysis of a human genome using next generation sequencing (NGS) can now cost the consumer under a thousand dollars3 its newly achieved cost effectiveness means that it can be integrated into molecular diagnostics workflows, despite its many challenges. The challenges of NGS include but are not limited to:
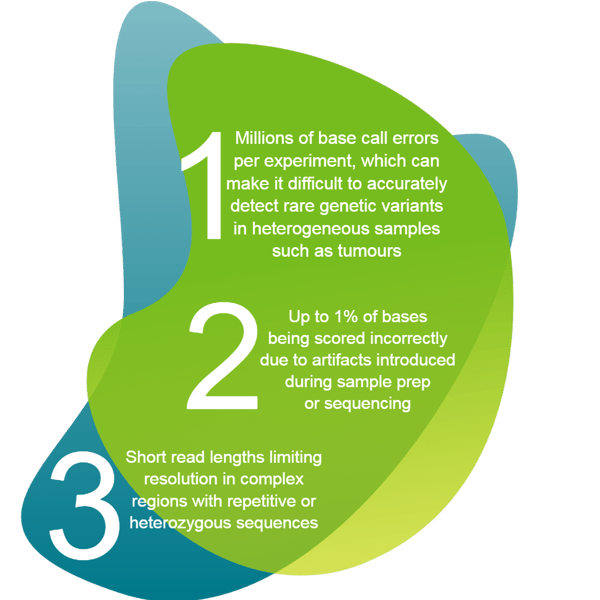
With so much room for error, the successful combining together of the different critical components in the NGS workflow represents a further challenge, whether for upstream sample prep, library construction, sequencing, or downstream bioinformatics analysis. This can be best achieved by automating as much of the process as possible, although the variable nature of NGS applications demands a more open automation approach.
From performing NGS on a tumour sample to determine the best course of treatment for an individual’s cancer chemotherapy, to using NGS-based techniques to trace and identify a pathogen at the start of a lethal epidemic, timing is critical as well, so the NGS workflow must be as efficient as possible, with fast turnarounds, of days rather than weeks. This article explains how some of the technical challenges associated with the use of NGS in molecular diagnostics can be effectively overcome.
NGS strategies: what is available?
There are at least three strategies to approach molecular diagnostics using NGS-based methods, and each of them come with their respective advantages and disadvantages. Our aim here is simply to highlight a few of their general characteristics. This is not an exhaustive list, and the individual applicability of the different methods will depend on the individual project and the resources available.1
- Panels analyse a limited number of genes associated with a specific group of diseases or phenotypes. These tests are especially indicated for well genetically described mendelian diseases comprising genes and mutations that have already been defined in the literature.
- Whole Exome Sequencing (WES) includes coding regions (exons) of all genes, corresponding to approximately 2% of the human genome. It is particularly useful when a panel cannot identify the cause of the disease, since it allows the detection of variants in candidate genes not covered by the panel. It is also indicated for polygenic phenotypes with several genes involved or those that are not associated with any previously described disease.
- Whole genome sequencing (WGS) covers almost completely all the information encoded in an individual’s DNA, including intronic and regulatory regions. One of the main advantages of WGS is identification of complex structural variations, such as copy number variations and chromosomal translocations, often at a resolution of a single nucleotide base. In addition, it allows the detection of mutations in genes that may not previously have been associated with a specific disease, even in non-coding regions.
Whichever strategy is used, success with NGS depends on a well-constructed library, with optimal yield and quality; minimal sequence bias; and low error rates. Starting with high yield and high-quality materials is the best thing you can do to ensure excellent NGS data.
The workflow and associated challenges described herein refer to “classical” short-read NGS, as opposed to Single-molecule real-time sequencing (SMRT), which generates long reads (upwards of 10K) for which requirements for DNA isolation as well as library prep and bioinformatics approaches are different. In all cases, the availability and level of completeness of the genome assembly and annotation will determine the applications and methods of choice.4
Challenge #1: Sample prep
Sample purification is critical to reliable NGS data, and the primary requirement for successful NGS is a nucleic acid template that is of adequate quality and purity. Problems can include low yields, poor quality, and difficult sample types, such as formalin-fixed, paraffin-embedded (FFPE) samples. If you are working on a genotyping project and would like to ship your DNA samples to LGC Biosearch Technologies, please ensure that you follow the guidelines on this page.
In cases where there may not be the time or budget in-house to optimise sample prep, a DNA/RNA purification service approach can also help in developing a fit-for-purpose solution for a specific project, specifically DNA and RNA extraction for research use only applications and cohort studies. Let Biosearch Technologies help with this step, by completing a short request form here.
Challenge #2: Library construction
The four main steps in preparing RNA or DNA for NGS analysis are generally:
(i) fragmenting and/or sizing the target sequences to a desired length
(ii) converting target to double-stranded DNA
(iii) attaching oligonucleotide adapters to the ends of target fragments, and
(iv) quantitating the final library product for sequencing.5
The size of the target DNA fragments in the final library is a key parameter for NGS library construction. Three approaches are available to fragment nucleic acid chains: physical, enzymatic, and chemical. DNA is usually fragmented using physical methods (acoustic shearing and sonication) or enzymatic methods.
A second post-library construction sizing step is commonly used to refine library size and remove adaptor dimers or other library preparation artefacts. Adapter dimers are the result of self-ligation of the adapters without a library insert sequence. These dimers use up space on the flow cell without generating any useful data. Magnetic bead-based clean up, or agarose gel purification can be used.
Consistent DNA shearing into appropriately sized DNA fragments is a critical step prior to starting DNA fragment library preparation. Biosearch Technologies has partnered with Diagenode, a leading manufacturer of water bath sonicators, to supply you with the best DNA shearing instruments for your NGS experiments, alongside fragmentation kits.
When considering DNA fragment library preparation, ready-optimised kits will reliably deliver complex libraries for your particular sample type, and the resultant library is more likely to be compatible with your downstream NGS sequencing protocol. The NxSeq® DNA library kits can be considered here.
There are many enzymes available for DNA and RNA library preparation. However, when looking for molecular diagnostics solutions, it is key to prioritise those that are manufactured using ISO 13485- and ISO 15189-accredited quality methods, and can provide consistent quality throughout scale-up, all the way through to commercialisation. This is a cost-effective way of driving high quality library preparation that enhances next generation sequencing accuracy and uniformity.
Contenders include but are not limited to:
- NxGen™ T4 DNA Ligase
- T4 Polynucleotide Kinase (PNK)
- Circligase™ ssDNA Ligase
- Circligase II ssDNA Ligase
- NxGen M-MuLV Reverse Transcriptase
- EpiScript™ RNase H- Reverse Transcriptase
- T4 RNA Ligase 2, Deletion Mutant
In the case of partnerships, Biosearch Technologies can also help you incorporate any enzyme of your choice into your specific NGS workflow.
Challenge #3: Sequencing and downstream analysis
Once both sample prep and library construction have been optimised, there are a number of sequencing platforms available, and there are numerous reviews on the subject.6 Despite all the developments in NGS however, the dramatic increase in NGS sequence throughput has come at a cost of much lower read accuracy and shorter sequence length compared with traditional Sanger sequencing.
This can be partially mitigated through increasing sequence coverage, and with bioinformatics, to overcome the obstacles associated with handling such massive datasets, and by developing tools to check sequence quality, conduct sequence alignment and assembly, and interpret the data.

Translating high volumes of short reads to diagnostic biomarkers
1. Images are analysed and converted into sequence reads using the manufacturer's base-calling system.
2. Reads are quality filtered and aligned, by de novo assembly or by mapping to a reference sequence - complete genome, subsets of a genome, a transcriptome or an exome.
3. Reads used to investigate genetic mechanisms underlying disease, via gene-expression profiling; SNPs novel transcripts or splice variants; detecting gene fusion and transcription factors, methylation status and histone modifications.
Given the inherently higher error rate in raw sequence data from NGS, one must also generate consensus benchmark datasets with reference samples.
Bioinformatics tools for analysing NGS data can be grouped into four general categories: base calling, alignment of reads to a reference, de novo assembly, and finally genome browsing, annotation and variant detection.
However, each NGS platform generates different types of data and often leads to different needs, and accurate alignment is mandatory so that NGS data can be considered biologically meaningful. NGS alignment is quite different from Sanger sequence alignment. Sanger data is already a cluster technology, and the read-out is already a consensus call, where next generation sequencing uses alignments to build a consensus call.
In addition, short read lengths, relatively high error rates in base calling and the sheer volume of NGS data make NGS alignment much more difficult than that for Sanger sequencing data. One limitation of aligning and assembling NGS short reads is that a large portion of them cannot be uniquely aligned to a reference when sequence reads are too short, and the reference is too complex.
Conventional alignment solutions such as the basic local alignment search tool (BLAST) and the BLAST-like alignment tool (BLAT) are efficient for aligning long reads, such as those generated by Sanger sequencing, but are inadequate for handling NGS short reads. To date, a variety of algorithms and software packages have been specifically developed for dealing with millions of NGS short reads. For example, MAQ and Bowtie are popularly used alignment tools.7
To help meet the challenge of bioinformatic and data analysis, Biosearch Technologies services can be used in a wide range of sequencing applications. These services are offered in combination with laboratory service projects for research use only, although many of the more recent NGS applications in human oncology are still in the clinical research phase. Learn about our expertise and how our services can help you get the most from your NGS data here.
Conclusion
With the cost of genome sequencing falling sharply, more and more personal genomes for individuals will become available. The new challenge will then be how to efficiently turn the large volumes of whole-genome sequence data into clinically useful information, including molecular diagnostics and treatment selection. At the other end of the spectrum, NGS will also become more readily used in pathogen detection and disease control.2
It is clear that NGS applications can yield useful and clinically relevant information for individual patients in clinical diagnostics.1 As databases for disease-specific mutations and pharmacogenomics become more comprehensive, NGS-based approaches will become more useful in clinical evaluation than traditional molecular diagnostic methods such as ELISA, that are usually used for one intended purpose, and NGS will indeed become a valuable and realistic tool in our fight to conquer global disease.8
In the video above, Emma Millican, Head of NGS at Biosearch Technologies, takes us through the importance of choosing the right components for NGS. She explains how an effective partnership between the scientist and the assay developer can help bridge the gap between biomarker discovery, to bring next generation molecular diagnostics assays efficiently to market.
Learn more about how Biosearch Technologies can help you meet the challenges for your own NGS projects, by subscribing to our blog below and checking out our NGS products and services.



References
- https://www.medscape.com/viewarticle/740460_3 (accessed 3 July 2019)
- https://www.clinlabint.com/detail/clinical-laboratory/molecular-diagnostics-of-pathogens-using-next-generation-sequencing/ (accessed 3 July 2019)
- https://www.forbes.com/sites/matthewherper/2017/01/09/illumina-promises-to-sequence-human-genome-for-100-but-not-quite-yet/ (accessed 3 July 2019)
- 4. Eid, J. et al. Real-Time DNA Sequencing from Single Polymerase Molecules, Science 323, 133-138 (2009). https://www.ncbi.nlm.nih.gov/pubmed/19023044 (accessed 3 July 2019)
- Head, S.R. et al, Library construction for next-generation sequencing: overviews and challenges. Biotechniques (2014) 56(2):61-4. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4351865/ (accessed 3 July 2019)
- Levy, S.E. and Myers, R. M. Advancements in Next-Generation Sequencing Annu. Rev. Genom. Hum. Genet. (2016) 17:95–115
- https://www.medscape.com/viewarticle/740460_3 (accessed 3 July 2019)
- Yolken H (1980) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2595839/ (accessed 3 July 2019)
