Updated : Wed, January 9, 2019 @ 5:03 PM
Genomic selection: 6 factors to consider when choosing between targeted GBS and microarrays
 Genomic selection through genotyping is more accurate than conventional breeding methods and promises to revolutionise crop and animal breeding. Gel-based technologies such as restriction fragment length polymorphism (RFLP) analysis and Sanger sequencing were used during the development of this field, followed by microarrays and PCR-based genotyping. Next generation sequencing (NGS) is now powering the development of more targeted genotyping by sequencing (tGBS) methods, including capture-based enrichment followed by analysis using NGS. The question is, which genotyping solution is right for the challenges you face? Let’s compare the main contenders, arrays and targeted genotyping by sequencing (tGBS), by looking at some key factors that will affect the efficiency of your breeding program.
Genomic selection through genotyping is more accurate than conventional breeding methods and promises to revolutionise crop and animal breeding. Gel-based technologies such as restriction fragment length polymorphism (RFLP) analysis and Sanger sequencing were used during the development of this field, followed by microarrays and PCR-based genotyping. Next generation sequencing (NGS) is now powering the development of more targeted genotyping by sequencing (tGBS) methods, including capture-based enrichment followed by analysis using NGS. The question is, which genotyping solution is right for the challenges you face? Let’s compare the main contenders, arrays and targeted genotyping by sequencing (tGBS), by looking at some key factors that will affect the efficiency of your breeding program.
Can you implement the flexible and scalable marker strategy you need?
The number of markers you need to screen for genomic selection depends on the species and the stage in your breeding cycle. Single nucleotide polymorphism (SNP) discovery involves 10,000–100,000 markers on perhaps as little as 5 samples, whereas the sweet spot for genomic selection is around 1,000–25,000 assays run on approximately 1,000 samples (see Figure 1). Being able to apply different levels of multiplexing using the same technology adds efficiency and consistency to your breeding program.
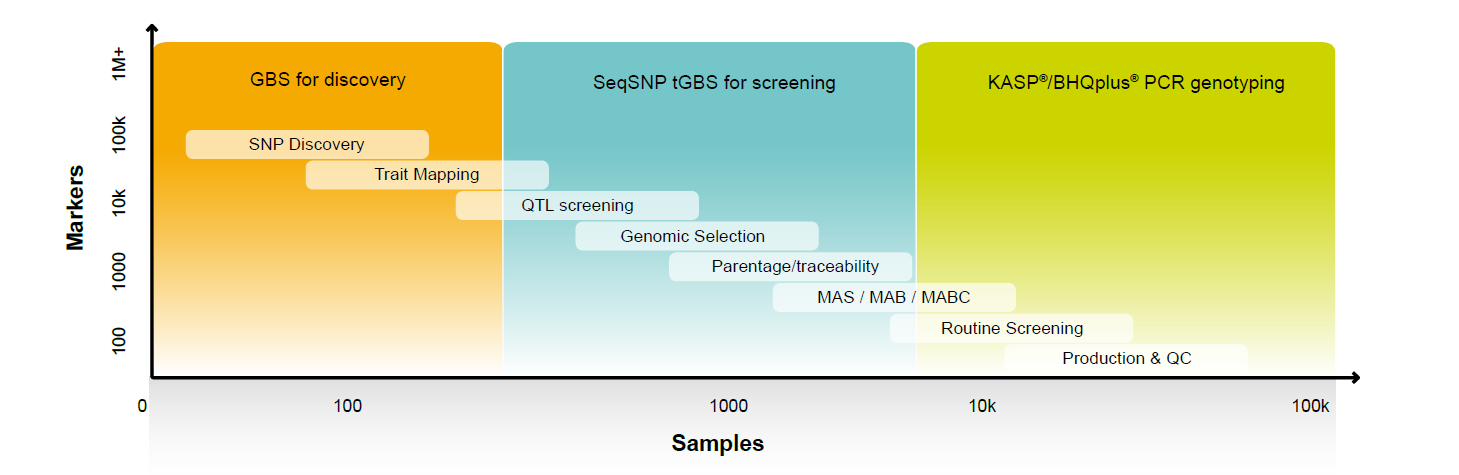
Figure 1. A typical breeding program involves moving from high coverage of a few samples in SNP discovery to medium multiplex levels for genomic selection.
Certainly arrays of different densities can deliver high and medium capacity SNP analysis, but this technology is very rigid, making it difficult to adapt marker density and composition based on the stage in your breeding program. There are, on the other hand, tGBS methods that can be used to screen up to 100,000 markers per sample but also function efficiently in that mid-plex sweet spot of 500 to 25,000 markers. This gives you the flexibility you need for genomic selection, even when you are working with multiple populations that have different genetic backgrounds.
Can you be cost-effective?
The effective application of genomic selection means screening a large number of samples quickly and efficiently, which can reduce breeding cycles by years. This speeds up time to market for new varieties, giving you that competitive edge. To achieve this requires the right technology and also cost efficiency. Array technology is lagging behind in terms of flexibility, and the high setup cost can also be daunting. On the other hand, data output and efficiency of NGS platforms is continually being improved, dramatically reducing the cost of NGS (Figure 2). Already today we can multiplex thousands of samples for tGBS on a single flow cell of even a medium throughput NGS system.
So basing sample selection on NGS analysis will inevitably drive up throughput while reducing costs. Added to that, highly efficient enrichment methods can reduce day-to-day operation costs even further.
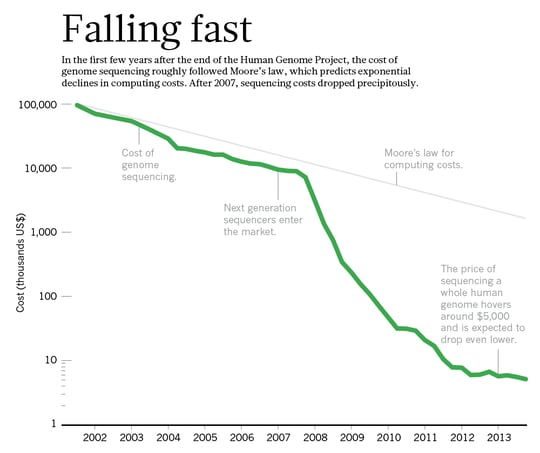
Figure 2. The cost of NGS is falling rapidly. Source: nature.com
Can you stay on target?
Using NGS for whole genome sequencing will deliver a relatively low cost per data point, but there are strong arguments for ensuring that analysis is limited to the specific genomic regions relevant to your study. For example, in most crop genomes, the exome corresponds to only 1–2% of the entire genome. Specifically targeting the regions of interest through capture and sequencing significantly reduces the cost of sequencing and data analysis (see 1).
Can you make the most of imputation?
One way to reduce genotyping cost is imputation, which is the statistical inference of unobserved alleles by using known haplotypes based on database information progenitors and sequenced parental lines. Imputation is cheaper in breeding programs because the numbers of markers that are used for screening are reduced. Therefore, accurate and informative imputation can make breeding strategies much more cost effective, but this can only be achieved with high-quality data from previously screened populations.
Imputation can be performed both from arrays and sequencing data. The trick is to select an optimized subset of existing markers. In the case of arrays, these design rounds can be very time consuming and prohibitively expensive. Added to that, it may be impossible to replace these markers since they are fixed on an array that may be the result of collaboration between many groups. In contrast, the lower setup costs and flexibility of tGBS make this approach much more attractive when developing imputation panels. With tGBS, any non-informative markers can be quickly and easily exchanged for others that may be more informative in further rounds of screening and imputation.
Does the technology fit into your breeding cycle?
The setup time for an array based on a new set of markers can be considerable, up to six months. In contrast, tGBS approaches can enable a turnaround time of less than 2 weeks, plus 4–6 weeks for the design of a new oligo library, which means you can fit it into a plant breeding cycle and improve selection of the accessions to be transplanted to the field and progressed. The result can be years of savings in development time.
Can you discover de novo variants?
Arrays discriminate targeted SNPs and are, by definition, fixed. Sequencing-based methods such as tGBS on the other hand enable the discovery of new SNPs and structural variants in flanking sequences of targeted SNPs. This increases the amount of genetic information you have at your fingertips, increasing the power of genomic selection. For example, in a study of 500 markers using sequences previously tested on an array, only 491 SNP sequences were originally selected to be common between the tGBS library and array data whereas tGBS discovered 5,733 de novo SNPs (2).
How to find the sweet spot with tGBS
As we have seen, exploiting genomic selection will help you produce new varieties faster. But it means finding a sequence-based genotyping solution that can meet your needs in terms of flexibility and cost-efficiency, while enabling you to carry out de novo SNP discovery, imputation, and much more. We will look at one way of achieving this in the last article in this series.
About the author: Darshna 'Dusty' Vyas
Dusty has been with LGC for the last 6 years working as a plant genetics specialist.
Her career began at the James Hutton Institute, formerly the Scottish Crop Research Institute, developing molecular markers for disease resistance in raspberries. From there Dusty moved on to Biogemma UK Ltd for a period of 13 years, where she worked primarily with cereal crops such as wheat, maize and barley. Through her participation in the Artemisia Project, funded by the Bill and Melinda Gates Foundation, at York University, she gained a vast understanding of the requirements by breeders for varietal development using molecular markers in MAS.
Dusty's goal is to further breeding programs for global agricultural sustainability using high throughput methods such as SeqSNP.
References
- Efficient genome-wide genotyping strategies and data integration in crop plants. Torkamaneh D et al. Theor Appl Genet. Mar;131(3):499–511 (2018)
- White paper: SeqSNP tGBS as alternative for array genotyping in routine breeding programs.
