Updated : Wed, December 14, 2022 @ 1:54 PM
When the polymerase chain reaction (PCR) was first described, the Klenow fragment of Escherichia coli DNA Polymerase I was the paramount enzyme for sequence extension. Due to its lack of stability at high temperatures, it must be replenished before each cycle.1 Upon the discovery of thermophilic bacteria which thrive at temperatures greater than 45 °C, heat-stable polymerases which function at higher temperatures were investigated in an effort to eliminate the need to replenish enzyme following each denaturation cycle.
 Taq DNA Polymerase was first isolated from the thermophilic bacterium Thermus aquaticus by Chien et al. in 1976.2 This extreme thermophile – originally discovered in the thermal springs of Yellowstone National Park in 19693 – has been a useful source of thermostable enzymes, the most known of which is Taq DNA polymerase (Taq). Taq can be isolated either from its original source or from its cloned gene expressed in E. coli. While many similarities in sequence and structure exist between E. coli DNA polymerase I and Taq, differences in enzyme functionality, character and dependencies make Taq the ideal polymerase for use in a variety of PCR applications, with the exception of sensitive measures of certain bacterial genes. This caveat is explained later.
Taq DNA Polymerase was first isolated from the thermophilic bacterium Thermus aquaticus by Chien et al. in 1976.2 This extreme thermophile – originally discovered in the thermal springs of Yellowstone National Park in 19693 – has been a useful source of thermostable enzymes, the most known of which is Taq DNA polymerase (Taq). Taq can be isolated either from its original source or from its cloned gene expressed in E. coli. While many similarities in sequence and structure exist between E. coli DNA polymerase I and Taq, differences in enzyme functionality, character and dependencies make Taq the ideal polymerase for use in a variety of PCR applications, with the exception of sensitive measures of certain bacterial genes. This caveat is explained later.
Characterisation of Taq DNA Polymerase
Size, activity and fidelity
While variable weights have been reported, the approximate size of Taq is 94 kDa,4 with the activity of a DNA polymerase localised to the C-terminus and 5′ to 3′ exonuclease activity localised to the N-terminus.5 Taq does not demonstrate the 3′ to 5′ exonuclease activity that has been observed in other polymerases, where a 3′-end mismatched base is excised during a “proofreading” process.6,7 The error rate – also known as the fidelity – is generally reported to be approximately 105 mutations per base per template doubling.8,9 One study found 98.8% of the errors to be base substitutions while only 1.2% were frameshift errors.8
Temperature dependency
Thermophiles are prevalent in nature and certain prokaryotic species thrive at temperatures above 45 °C. The temperature-dependency of Taq makes it optimum at 75-80 °C, where its catalytic activity can reach 150 nucleosides per second.4
Thermostability
Taq is heat stable, making it compatible with hot-start protocols, but at temperatures over 90 °C the half-life drops rapidly. The half-life is 40 minutes at 95 °C, but drops to only 5-6 minutes at 97.5 °C.10 As a consequence, PCR protocols with a denaturing step at 95 °C are likely to denature a proportion of the Taq present.
Monovalent and divalent cation dependencies
Monovalent and divalent ions, such as K+ and Mg2+, are required to neutralise the negative charges on the backbone of DNA. This reduces the repulsive interactions between the negatively charged DNA strands, enabling polymerisation to proceed.11 These cations are usually added in their salt forms – KCl and MgCl2, respectively.
Potassium salt (KCl) is a common component of Taq buffers. The optimum concentration of KCl is generally around 50 mM, however, reducing the concentration can be beneficial for generating long PCR products. For shorter products below 1,000 base pairs, an increase in KCl concentration increases specificity by reducing the efficiency of longer stranded DNA fragments separating.12
Taq, like other polymerases, depends upon the presence of divalent cations, namely Mg2+ or Mn2+, to act as a cofactor during polymerisation.4,13 An increase in Mg2+, the most commonly used cofactor, leads to an increase in product yield, but a reduction in the specificity and fidelity.12 The optimum concentration of MgCl2 is approximately 2 mM.4
Taq requires the presence of all four species of deoxyribonucleoside triphosphates and DNA for optimum catalytic activity.
pH dependency
The pH optimum for the enzyme is within the range of 8-9.4 pH units,2,4 and will vary depending on the buffer system used. Common commercially available Taq buffers are typically in this range and will often contain 10-50 mM Tris-HCl for pH stability during PCR.
Heat-activation
Hot start PCR is a method used to reduce non-specific DNA amplification and the formation of primer dimers that are thought to occur during the room temperature conditions of sample preparation.14 A hot start approach can be achieved through several methods, including inhibiting Taq using chemical modifications, antibodies, protein fusions and aptamers. Enzyme activity remains blocked until the initial denaturation temperature is reached. Alternatively, essential components of the reaction can be physically separated from Taq until the annealing temperature is reached.14
Hot start activation is particularly beneficial when multiple pairs of primers are being used, when the template DNA is complex, or when there are fewer than 104 copies of template DNA present.15
Applications of Taq
Taq is the enzyme of choice for most PCR applications and is used widely in molecular diagnostics.
The inherent 5′ to 3′ exonuclease activity of Taq means that PCR amplification can be achieved simultaneously with signal release from a target-specific fluorogenic probe. The 5′ to 3′ exonuclease activity of Taq cleaves the 5′ terminus of a hybridised oligo probe to release both mono- and oligonucleotides. The probe is hydrolysed concomitant with strand replication so that the accumulating fluorescent signal correlates with amplification.16 This enables the use of PCR with dual-labelled probes for applications such as pathogen detection, SNP genotyping and gene expression analysis.
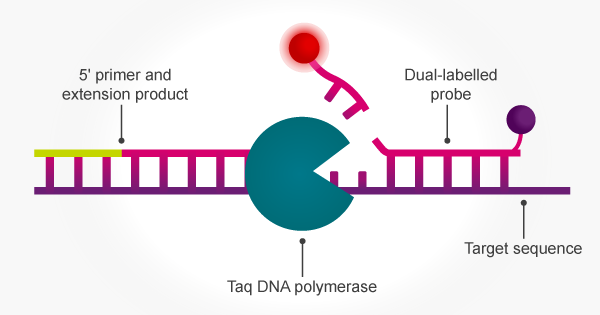
The great caveat: uninvited guests
It has been well reported that Taq often has contaminating bacterial DNA, possibly carried over from the expression vector system or other sources used during polymerase manufacture.17,18 This residual contamination may limit the use of Taq in the detection of dilute bacterial DNA in certain samples. Rodent DNA has also been found as a contaminant, with the authors of a study concluding that this was likely due to mouse monoclonal antibodies being used for hot start activation of Taq.19
In 2021, a study investigated 16 commercial Taq DNA polymerases for DNA contamination. The authors found 11 out of 16 Taq polymerases were contaminated with beta-lactamase antibiotic resistance genes and 15 of the 16 Taq polymerases contained 16S rRNA. This is significant as antibiotic resistance genes are frequently used as selection markers for GMOs and 16S rRNA is often used as a positive control in PCR.20
Several methods for removing contaminating DNA from Taq have been tested and appear in the literature:
Exposure to ultraviolet light causes DNA to form pyrimidine dimers,21 therefore making DNA resistant to amplification. Long wave UV light has been used for decontamination but has been found to also affect the integrity of the Taq polymerase, reducing the efficiency of nucleoside incorporation. Corless et al. found UV irradiation resulted in a 4-log reduction in PCR sensitivity, which was equivalent to increasing the qPCR quantification cycle (Cq) by 12.22
DNase treatment to digest DNA in Taq preparations has been shown to eliminate contamination.22,23 However, the DNase subsequently requires heat inactivation which is likely to reduce the activity of Taq,23 and residual DNase from incomplete inactivation may be responsible for reduced PCR sensitivity.22 DNase treatment can also be used in combination with restriction endonucleases, although these introduce the risk of adding contaminants from their own manufacture22 and can also inhibit PCR.23
Serial dilution of Taq (up to 32-fold) to dilute-out contaminating bacterial DNA was found to maintain activity and sensitivity, although the reaction plateaued at lower cycle numbers. This effect generates a lower signal in end-point analysis, with minimal consequence to quantification based on a threshold during the exponential phase.24
Treatment with nylon membrane disks for 24 hours was found to remove contaminating DNA as it adsorbed onto the disks. Furthermore, It did not decrease the Taq activity.20
DNA contamination of Taq and other PCR reagents is particularly problematic where very sensitive detection is employed, such as in digital droplet PCR. As such, careful consideration must be taken during assay design and optimisation.
Learn more about controlling contamination and ensuring reliable qPCR results.

References
- Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H. Specific Enzymatic Amplification of DNA In Vitro: The Polymerase Chain Reaction. Cold Spring Harb Symp Quant Biol. 51:263-273. https://doi.org/10.1101/SQB.1986.051.01.032. Published 1986. Accessed 10 February 2022.
- Chien A, Edgar DB, Trela JM. Deoxyribonucleic acid polymerase from the extreme thermophile Thermus aquaticus. J Bacteriol. 127(3):1550-1557. https://doi.org/10.1128/jb.127.3.1550-1557.1976. Published 1976. Accessed 10 February 2022.
- Brock TD, Freeze H. Thermus aquaticus gen. n. and sp. n., a nonsporulating extreme thermophile. J Bacteriol. 98(1):289-297. PMID: 5781580. Published 1969. Accessed 10 February 2022.
- Lawyer FC, Stoffel S, Saiki RK, Chang S-Y, Landre PA, Abramson RD, Gelfand DH. High-level Expression, Purification, and Enzymatic Characterization of Full-length Thermus aquaticus DNA Polymerase and a Truncated Form Deficient in 5′ to 3′ Exonuclease Activity. PCR Methods Appl. 2(4):275-87. https://doi.org/10.1101/gr.2.4.275. Published 1993. Accessed 8 February 2022.
- Longley MJ, Bennett SE, Mosbaugh DW. Characterization of the 5′ to 3′ exonuclease associated with Thermus aquaticus DNA polymerase. Nucleic Acid Research. 18(24):7317-7322. https://doi.org/10.1093/nar/18.24.7317. Published 1990. Accessed 11 February 2022.
- Denomme GA, Rios M, Reid ME. Molecular Protocols in Transfusion Medicine. Part 2 - General Considerations. Academic Press. https://doi.org/10.1016/B978-012209370-8/50003-9. Published 2000. Accessed 11 February 2022.
- Park Y, Choi H, Lee DS, Kim Y. Improvement of the 3′ – 5′ Exonuclease Activity of Taq DNA Polymerase by Protein Engineering in the Active Site. Mol. Cells. 7(3):419-424. PMID: 9264032. Published 1997. Accessed 11 February 2022.
- Potapov V, Ong JL. Examining Sources of Error in PCR by Single-Molecule Sequencing PLoS One. 12(1). https://doi.org/10.1371/journal.pone.0169774. Published 2017. Accessed 11 February 2022.
- McInerney P, Adams P, Hadi MZ. Error Rate Comparison during Polymerase Chain Reaction by DNA Polymerase. Mol Biol Int. 2014:287430. https://doi.org/10.1155/2014/287430. Published 2014. Accessed 11 February 2022.
- Van Pelt-Verkuil E, van Belkum A, Hays JP. Principles and Technical Aspects of PCR Amplification. Taq and Other Themostable DNA Polymerases. Springer. https://doi.org/10.1007/978-1-4020-6241-4. Published 2008. Accessed 14 February 2022.
- Tan ZJ, Chen SJ. Nucleic acid helix stability: effects of salt concentration, cation valence and size, and chain length. Biophys J. 90(4):1175-1190. https://doi.org/10.1529/biophysj.105.070904. Published 2006. Accessed 14 February 2022.
- Lorenz TC. Polymerase chain reaction: basic protocol plus troubleshooting and optimization strategies. J Vis Exp. (63). https://doi.org/10.3791/3998. Published 2012. Accessed 11 February 2022.
- Stengel G, Purse BW, Kuchta RD. Effect of transition metal ions on the fluorescence and Taq-catalyzed polymerase chain reaction of tricyclic cytidine analogs. Anal Biochem. 416(1):53-60. https://doi.org/10.1016/j.ab.2011.04.033. Published 2011. Accessed 14 February 2022.
- Paul N, Shum J, Le T. RT-PCR Protocols: Hot Start PCR Methods. Molecular Biology (Methods and Protocols), vol 630. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-60761-629-0_19. Published 2010. Accessed 8 February 2022.
- Green MR, Sambrook J. Hot Start Polymerase Chain Reaction (PCR). Cold Spring Harb Protoc. (5). https://doi.org/10.1101/pdb.prot095125. Published 2018. Accessed 8 February 2022.
- Holland PM, Abramson RD, Watson R, Gelfand DH. Detection of specific polymerase chain reaction product utilizing the 5′ – 3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. 88:7276-7280. https://doi.org/10.1073/pnas.88.16.7276. Published 1991. Accessed 10 February 2022.
- Masalov YK, Heydarov RN, Shashkov IA, Chebotar IV, Zasedatelev AS, Mayansky NA, Mikhailovich VM. Exogenous contaminating DNA in Taq polymerases: A method to avoid false-positive results when detecting the blaTEM gene. Journal of Microbiological Methods. 160:36-41. https://doi.org/10.1016/j.mimet.2019.03.018. Published 2019. Accessed 8 February 2022.
- Maiwald M, Ditton H-J, Sonntag H-G, von Knebel Doeberitz M. Characterization of contaminating DNA in Taq polymerase which occurs during amplification with a primer set for Legionella 5S ribosomal RNA. Molecular and Cellular Probes. 8(1):11-14. https://doi.org/10.1006/mcpr.1994.1002. Published 1994. Accessed 8 February 2022.
- Witt N, Rodger G, Vandesompele J, Benes V, Zumla A, Rook GA, Huggett JF. An assessment of air as a source of DNA contamination encountered when performing PCR. J Biomol Tech. 20(5):236-240. PMCID: PMC2777341 Published 2009. Accessed 8 February 2022.
- Zhao SR, Qu B, Kim JK, Lee B. Decontamination of DNA in Taq DNA polymerase reagents using nylon membranes for monitoring of GMOs. Plant Biotechnol Rep 15, 783–790 (2021). https://doi.org/10.1007/s11816-021-00723-z. Published 2021. Accessed 8 February 2022.
- Kiefer J. Chromosomal Alterations. Effects of Ultraviolet Radiation on DNA. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-540-71414-9_3. Published 2017. Accessed 10 February 2022.
- Corless CE, Guiver M, Borrow R, Edwards-Jones V, Kaczmarski EB, Fox AJ. Contamination and sensitivity issues with a real-time universal 16S rRNA PCR. J Clin Microbiol. 38(5):1747–1752. https://doi.org/10.1128/JCM.38.5.1747-1752.2000. Published 2000. Accessed 10 February 2022.
- Klaschik S, Lehmann LE, Raadts A, Hoeft A, Stuber F. Comparison of different decontamination methods for reagents to detect low concentrations of bacterial 16S DNA by real-time PCR. Molecular Biology. 22(3):231-242. https://doi.org/10.1385/MB:22:3:231. Published 2002. Accessed 10 February 2022.
- Spangler R, Goddard NL, Thaler DS. Optimizing Taq polymerase concentration for improved signal-to-noise in the broad range detection of low abundance bacteria. PLoS One. 4:(9). https://doi.org/10.1371/journal.pone.0007010. Published 2009. Accessed 10 February 2022.
